
What are lysosomal storage disorders?
About lysosomal storage disorders
Lysosomal storage disorders (LSDs) are a group of rare genetic conditions that result in an accumulation of undegraded substrates in lysosomes. There are more than 70 different types of LSDs, some of which have subtypes. These conditions are lifelong, usually get worse over time (are progressive), and affect several organs. They can be serious and sometimes shorten lifespan, but for many patients the disease can be managed.1,2
Three things to know

Rare does not mean alone
The combined incidence rate of LSDs worldwide is up to 1 in 5,000 to 1 in 8,000 live births1
Symptoms can start early in life
Most people with LSDs begin to have symptoms in infancy or childhood1
Diagnosis can be key
For many patients, diagnosis is helpful for managing their LSD1
.
What causes lysosomal storage disorders?
The majority of LSDs are caused by pathogenic variants on certain genes that code for lysosomal enzymes. These pathogenic variants cause a deficiency in the lysosomal enzyme or impact their ability to function properly. When that happens, naturally-occurring substances that our bodies usually break down build up—sometimes to toxic levels. That’s why LSDs are known as enzyme deficiency diseases. LSDs are also inherited, which means the genes that cause them can be passed down in families.1
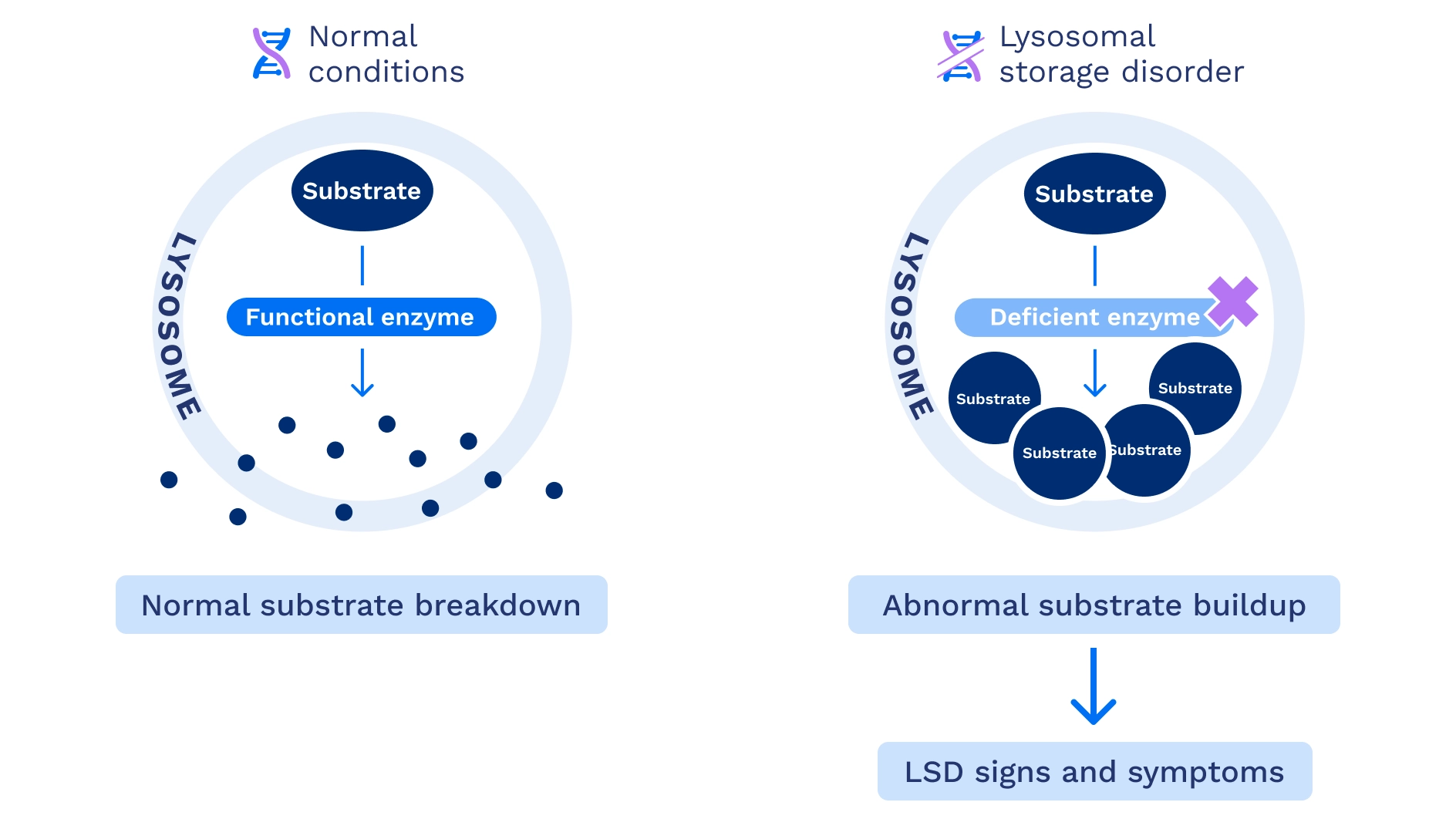
What are lysosomes and enzymes?
Lysosomes and enzymes play key roles in metabolism. Lysosomes are small, sac-like structures found in almost every cell of the body. Lysosomes contain enzymes that are responsible for breaking down substrates and act as a cell's recycling center. In LSDs, the material that should be broken down builds up instead. This material is called the substrate and in many cases is a type of fat or sugar.1,3
Enzymes are proteins inside lysosomes that can break down specific substrates. An LSD is usually caused by changes in a single gene, which limits the activity of a certain enzyme. As a result, the substrate builds up, leading to the signs and symptoms of the LSD.1
Types of lysosomal storage disorders
There are approximately 70 LSDs identified, some of which have subtypes. Most happen because a specific enzyme is deficient or doesn’t work well (and the exact enzyme differs per disease). LSDs are grouped into categories based on the kind of substrate—like fat or sugar—that builds up.1
Some LSD categories include:
- Sphingolipidoses, caused by buildup of a form of fatty substance
- Glycogen storage diseases, caused by buildup of glycogen, a form of sugar
- Mucopolysaccharidoses, caused by buildup of more complex sugar molecules
Specific LSDs can include Fabry disease, Tay-Sachs disease, Gaucher disease, mucopolysaccharidosis type I (MPS I), acid sphingomyelinase deficiency (ASMD) disease (also called Niemann-Pick disease), and Pompe disease, among others.1
Symptoms of lysosomal storage disorders
The substrates that build up in LSDs can impact different organs. Symptoms can be different depending on the LSD, and usually get worse over time, which means the disease is progressive. In some conditions, symptoms show up soon after birth, while with others, symptoms may not appear until childhood or even adulthood.1
What are the common symptoms of lysosomal storage disorders?
Some symptoms* that may occur in LSDs include:
- Delays in intellectual and/or physical development
- Mobility, vision, and/or hearing issues
- Difficulty breathing
- Bleeding or bruising easily
- Enlarged organs, such as liver or spleen
- Joint stiffness and pain
- Bone deformities
- Fatigue
*These are not all of the possible symptoms for LSDs, and not all patients will experience those listed. Be sure to talk to your doctor about any symptoms that may concern you.
How are LSDs diagnosed?
Tests for LSDs depend on the specific disease, but usually include blood tests to look for genetic variants or enzyme levels, and certain scans such as X-rays, MRIs, or EKGs. LSDs can be diagnosed at all stages of life. Some people may find out they have an LSD because they experience certain symptoms, while others can get a diagnosis after someone in their family has undergone genetic testing.1
Video: Understand the foundations of a family tree
Video: Understand the foundations of a family tree
LSDs run in families
It’s important to understand how LSDs can run in families. One helpful way to figure out who might be at risk for having the disease, is to look at your family history. You can work with a genetic healthcare provider or a CareConnect Patient Education Liaison (PEL) to create a visual representation of your family history called a family tree. Contact us at 1-800-745-4447 or email info@CareConnectPSS.com.
Screening for LSDs
Sometimes, children are diagnosed a few days after birth as a result of newborn screening. While all babies in the United States are screened for various conditions at birth, only certain states test for specific LSDs. LSDs can affect people of many different races and ethnicities, though people with certain backgrounds, like Ashkenazi Jewish heritage, may be at greater risk for LSDs.4,5 To learn more about screening options in your state, speak with your doctor.
The importance of testingSince LSDs can run in families,1 it's important to get tested as soon as possible if you or a family member think you may have the disease. Talk to your doctor about your options for testing. |

Treatment for LSDs
There are currently no cures for LSDs, but there are treatments and approaches available to help manage the condition. Each type of LSD can have different treatments.1 Your healthcare provider and care team will determine which treatments might be right for you.
Support for people living with an LSD
CareConnect offers personalized support services for eligible patients and families living with certain LSDs (ASMD, Fabry, Gaucher type 1, MPS I, and Pompe disease). An experienced team is ready to provide you with individualized support.
Call 1-800-745-4447 (for English, press 3; para español, oprima 7) or email info@CareConnectPSS.com.

References
1 Rajkumar V, Dumpa V. Lysosomal Storage Disease. StatPearls. 2025.
2 Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. Oct 1 2018;4(1):27. doi:10.1038/s41572-018-0025-4
3 Cooper G. Lysosomes. The Cell: A Molecular Approach. 2nd ed. Sunderland (MA): Sinauer Associates; 2000.
4 Risch N, Tang H, Katzenstein H, Ekstein J. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet. Apr 2003;72(4):812-22. doi:10.1086/373882
5 About Newborn Screening. United States Newborn Screening Information Center (NBSIC): Health Resources & Services Administration. Accessed August 25, 2025 from: https://newbornscreening.hrsa.gov/about-newborn-screening.
MAT-US-2511948-v1.0-12/2025