Living with MPS I disease
Mucopolysaccharidosis (mew-koh-poli-sak-er-I-doh-sis) type I (MPS I) is a lifelong condition, which means everyone who is affected by MPS I is born with it, even if they don’t show signs right away. Most people with MPS I begin to have symptoms early in life, but the condition can affect people of any age, male or female.1,2
MPS I is a rare condition—the severe form (Hurler syndrome) occurs in an estimated 1 in 100,000 births and the attenuated (less severe) form (Hurler-Scheie or Scheie syndrome) occurs in around 1 in 500,000 births.2
Living with MPS I is a unique experience for each individual affected by the condition. Even though MPS I can affect everyone differently, it’s important to remember that you and your loved ones are not alone. Learning more about MPS I and the support available can help you feel more empowered to take control of your healthcare journey whether it’s you or your child who has MPS I.

How does MPS I affect the body?
Many different organs and systems within the body can be impacted when a person has MPS I. Understanding more about how the condition affects the body can help you understand why certain symptoms may occur.
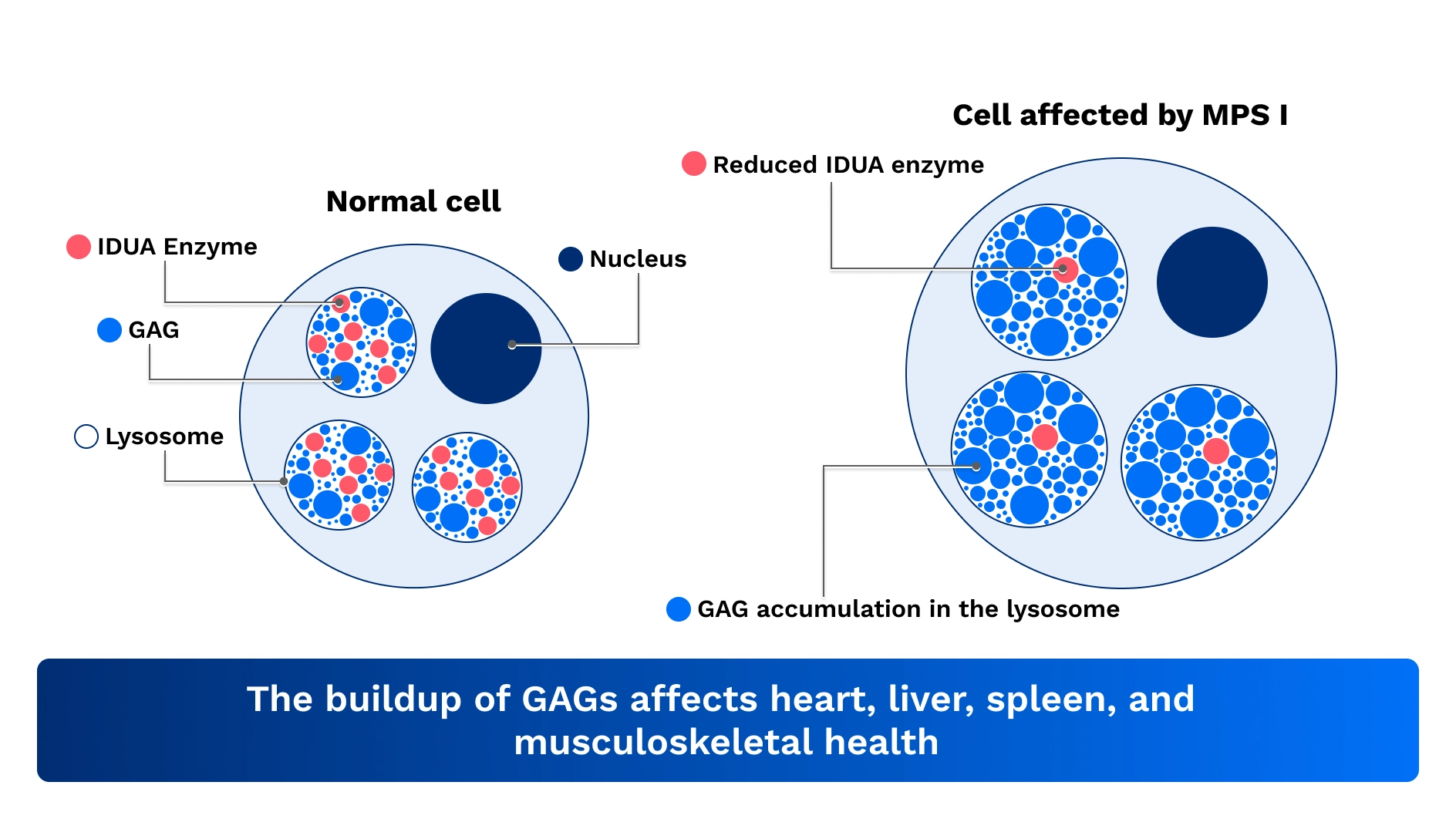
MPS I is a type of condition called a lysosomal storage disorder. Lysosomes are small sacs inside cells that contain enzymes to break down waste materials in the body. When the enzymes inside lysosomes don’t work properly, waste can build up in different organs, causing damage and leading to signs and symptoms of the condition.
When a person has MPS I, changes in the IDUA gene called pathogenic variants prevent the body from being able to produce enough functional alpha-L-iduronidase, or IDUA enzyme. The IDUA enzyme helps your body break down and remove substances called glycosaminoglycans, or GAGs. GAGs are a type of sugar that help make bones, cartilage, skin, tendons, and other tissues.
When the body is unable to make enough functional IDUA enzyme, excess GAGs build up inside the cells over time and lead to signs and symptoms seen in MPS I. Organs and tissues affected by this buildup can include the heart, liver, spleen, musculoskeletal system, and in more severe cases, the brain. For children who are affected by MPS I, the condition might also affect their growth and development.1-3
How is MPS I managed and monitored?
Because MPS I can get worse over time, monitoring is important for helping manage the condition and slowing progression. Not everyone who is affected will have the same symptoms or experience living with MPS I.1,2
It is important to work with your doctor to monitor your signs and symptoms carefully for any changes, so your care can be adjusted if needed.
What kind of doctors treat MPS I?
Since many different organs and systems in the body can be affected by MPS I, care plans often involve a team of healthcare specialists to ensure all symptoms are managed and monitored adequately.1,2
Your MPS I multidisciplinary care team may include:
- Audiologist – assesses hearing health, including potential hearing loss
- Cardiologist – assesses heart health, including function of valves
- Gastroenterologist – assesses for hernias and other digestive issues
- Genetic counselor – helps affected individuals and their families understand their genetic status and make informed treatment decisions
- Metabolic disease specialist – treats inherited conditions of metabolism
- Neurologist – assesses brain, spinal cord, and nerve health
- Ophthalmologist (eye doctor) – assesses changes in vision, corneal clouding, and other eye issues
- Orthopedist – specializes in monitoring and treating joint and skeletal issues
- Otolaryngologist (ENT) – specializes in airway support
- Pulmonologist – assesses respiratory health, assists with sleep studies
- Social worker – assists with coping, support, and resources
Your primary care physician or your child’s pediatrician can help coordinate care among specialists, based on your personalized needs.

Assessments used to monitor MPS I
Your doctor may want to repeat certain assessments periodically to help keep track of the condition and to help guide your care plan for MPS I. Examples of some possible assessments that may be used are provided below.1
Every 6 months
- General health and updated medical history
- Physical exam
- Growth monitoring (including weight and height)
- General appearance
- Lungs and breathing
- Vital signs (such as blood pressure)
- Laboratory tests (including GAG levels in the urine)
Every 12 months
- Ears and hearing
- Eyes and vision
- Sleep study
- Developmental test (such as IQ test)
Every 2 years
- CT scans or MRI imaging of brain and spinal cord
- Nerve signal testing
- Heart function
- X-ray to check bones and joints
- Liver and spleen size measurement
Please note that this is not a comprehensive list of all possible assessments for MPS I. Your doctor will decide what assessments should be done and how often they need to be made based on your personalized medical needs.
Self-monitoring for MPS I
You can also monitor your, or your child’s, MPS I by keeping a record of any changes in your existing symptoms or any new ones that occur.
Keeping track of symptoms can make it easier for your healthcare team to monitor your progress. Your care team can then decide if the condition is being managed, or if any changes are needed to your care plan.1
What are the different forms of MPS I?
MPS I can be classified as a severe form or an attenuated (less severe) form, based on the age of onset, severity of symptoms, rate of progression, and whether there is any early and direct involvement of the brain.1,2
Hurler syndrome
(severe form)
Hurler syndrome is named after the doctor who first discovered it, and is the most common form of MPS I. People who experience this form have symptoms that advance fast, damage to the brain and other key organs, and shortened lifespan.1,2,4
- Neurologic symptom severity: Significant mental delay with loss of acquired skills
- First appearance of symptoms: Early infancy (around 6 months)
- What it affects: Brain, bones, and key organs including heart, lungs, liver, and spleen
- Worsening over time: Rapid
- Life expectancy (without disease management): Less than 10 years
Hurler-Scheie syndrome
(attenuated form)
Hurler-Scheie syndrome describes the condition where symptom severity and worsening over time are between Hurler and Scheie syndromes. People with this form have symptoms that advance slower than Hurler, but faster than Scheie. They may experience normal brain development or slight development delay. Lifespan is between that of Hurler and Scheie.1,2,4
- Neurologic symptom severity: No/mild mental delay; learning disabilities
- First appearance of symptoms: Infancy to childhood (around 2 years)
- What it affects: Brain, bones, and key organs including heart, lungs, liver, and spleen
- Worsening over time: Variable
- Life expectancy (without disease management): 20-30 years
Scheie syndrome
(attenuated form)
- Scheie syndrome is an attenuated (less severe) form of MPS I. People with this condition present with symptoms that worsen more slowly over time, no damage to the brain, and a longer lifespan (into adulthood) compared to Hurler syndrome.1,2,4
Neurologic symptom severity: None - First appearance of symptoms: Childhood (5 years)
- What it affects: Bones and key organs including heart, lungs, liver, and spleen
- Worsening over time: Variable to slow
- Life expectancy (without disease management): Adulthood
.
How is MPS I inherited?
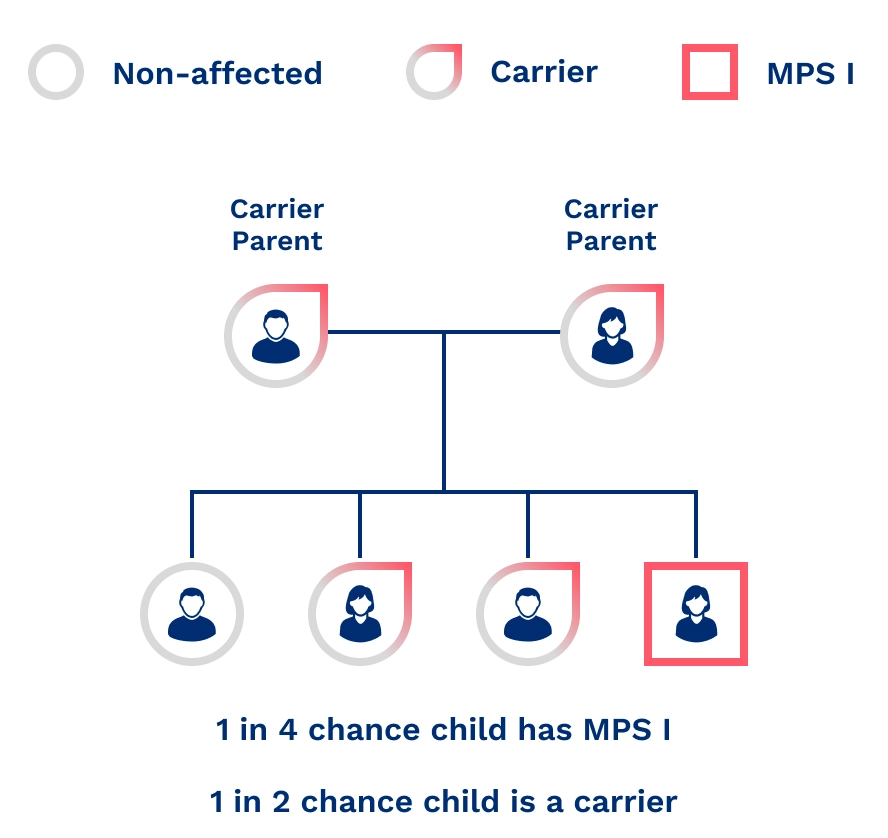
MPS I is a genetic condition caused by changes (also known as pathogenic variants) in the IDUA gene. It is inherited in an autosomal recessive pattern, which means a person needs to inherit two altered copies of the gene—one from each biological parent—to have the condition. Males and females are equally likely to inherit copies of the affected gene.2
People with only one altered copy are called carriers. Carriers do not have MPS I, but they can still pass the altered gene to their children.2
The likelihood of MPS I being passed down to children depends on the genetic status of the parents.2 When both parents are carriers, there is:
- A 25% chance that their child will have MPS I
- A 25% chance that their child will not have MPS I or be a carrier
- A 50% chance that their child will be a carrier of MPS I, without having the condition
These chances remain the same for every pregnancy, no matter if previous children were affected.
The importance of family screening and genetic testing
Since MPS I can run in families, if one person is diagnosed with MPS I, relatives should consider talking with their doctor or a genetic counselor about family screening or possibly getting tested.2
- Genetic counselors can help you understand the inheritance pattern of MPS I and how this relates to your genetic status. A genetic counselor can advise you on who may be at risk of being a carrier or having MPS I in your family, as well as whether the wider family should be informed. Genetic counselors can help you make informed medical and personal decisions based on your risks, including planning for pregnancy.
- Family screening can help your family members quickly find out if they have MPS I or are a carrier of the affected gene.
More information about living with MPS I
If you are or someone close to you is affected by MPS I and looking for more information, additional resources, or support, contact our CareConnect team Monday through Friday, 8AM-6PM EST.
Call 1-800-745-4447 (for English, press 3; para español, oprima 7) or email info@CareConnectPSS.com.

References
1 Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on M, Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. Jan 2009;123(1):19-29. doi:10.1542/peds.2008-0416
2 Clarke LA. Mucopolysaccharidosis Type I. 2002 Oct 31 [Updated 2025 Dec 4]. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Accessed January 7, 2026. https://www.ncbi.nlm.nih.gov/books/NBK1162/.
3 De Ponti G, Donsante S, Frigeni M, et al. MPSI Manifestations and Treatment Outcome: Skeletal Focus. Int J Mol Sci. Sep 22 2022;23(19)doi:10.3390/ijms231911168
4 Beck M, Arn P, Giugliani R, et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. Oct 2014;16(10):759-65. doi:10.1038/gim.2014.25
MAT-US-2602116-v1.0-03/2026